

Online computational tools, e.g. webservers, have been used extensively for research analysis. Many of such webservers require user manual inputs that can be laborious. In such cases, it is possible to automate some tasks using scripts, especially when performing similar tasks with multiple inputs. This article describes one such script, using Python programming language, for the webserver AlloSigMA for the analysis of allosteric communications in proteins. Our Python script can minimize human inputs and reduces human errors in the use of the AlloSigMA server.
Computational biology has come a long way with multiple purposes for investigation into biomedical data. Some examples of this include OMICTools and ExPASy (Artimo et al., 2012; Henry et al., 2014) as resource hubs for scientists to select bioinformatics tools and perform analyses with applications in research areas such as drug design and immunology. One reason for the convenience of bioinformatics tools is that they are often available as webservers, for example BLAST (Altschul et al., 1990) and ClustalW (Thompson et al., 1994) for sequence analysis, SWISS-PROT (Bairoch & Boeckmann, 1991) for protein structural analysis, alongside many others that include specialized areas of interest.
One such specialized area of interest is the field of allostery, which involves the analysis of distal communication effects across regions of a protein. Computational study of allosteric signaling have assisted in several experimental observations (Kurochkin et al., 2017; Zhao et al., 2017; Su et al., 2018; Zhao et al., 2018) to further unravel the relationship between protein structure and functions. One example is the AlloSigMA server (Guarnera et al., 2017). The back-end algorithm adopts a structure-based statistical mechanical model to quantify allosteric effects in protein structures in ligand binding and/or mutations (Guarnera & Berezovsky, 2016). Working with the server, users can interactively select the residues to simulate the binding event (i.e. choosing “SITE”) or mutation event (i.e. choosing “UP-MUT” or “DOWN-MUT”). The corresponding results will be instantly retrieved in a user-friendly graphical demonstration of allosteric communication within the protein target. However, this does not apply for single point mutation scanning.
In the case of mutational scanning, manual entry of mutational residues has to be performed (Su et al., 2017; Chiang et al., 2018). However, such manual entry poses challenges in large systems. For example, a number of members took part in the process of generating mutational data using AlloSigMA manually in one of our previous work utilizing the server (Su et al., 2018).
To address this manual task of submission and retrievals of single mutation scanning, which could involve artifacts, we presented an automated submission-retrieval python script for the interactive webserver. The idea to initiate our script is similar to the concept of automating in the MODFLOW model (Bakker et al., 2016) for data collection and processing by implementation of a python language script to its graphic user interface.
The script was written using Python 3.5.2 (Guido, 1995), as shown in Figure 1. We have chosen to use Python scripting for various reasons. Python is generally a high-level programming language, making it instinctual and user-friendly (Lehrer, 2014). Furthermore, Python is also considered a powerful tool (Oliphant, 2007) with thousands of scientific open source libraries, such as SciPy (Jones et al., 2014), NumPy (Oliphant, 2006), Pandas (McKinney, 2010) and Matplotlib (Hunter, 2007), some of which are applied in our script.
We elaborate some essential elements below:
The Selenium Python library was used as the main imported library, including WebDriver for website navigation. Since the Selenium library is unable to communicate directly with the web browsers, the drivers act as a medium for Selenium software to parse the script commands to the web browsers. Different drivers are required for different web browsers, e.g. Chromedriver and Geckodriver are drivers for Chrome and Firefox, respectively.

Figure 1: Pseudocode of the automated submission script to AlloSigMA server
To demonstrate how the script works, we performed mutational scanning on T4 Lysozyme on the AlloSigMA server using the script.
T4 Lysozyme (Enterobacteria phage, PDB: 253L) was chosen as the target due to its sufficient small size (164 residues) for the optimal processing time as well as its well-studied properties, e.g. structural stability, allosteric effects provided with numerous experimental structural data and characterized mutants (Shoichet et al., 1995; Sinha & Nussinov, 2001). For example, the T4 Lysozyme mutants (mutations at position P86) exhibited reduced catalytic activity (Alber et al., 1988). The T4 lysozyme active site includes residues Glu11 and Asp20 and the substrate binding site includes residues Ser117 and Asn132.
In this analysis, we sought to investigate the allosteric signaling propagating across the protein structure, in particular, the effects on the active site and the substrate-binding site. To do so, we performed the perturbation at each single residue and detected the responses at the two mentioned sites hence quantifying the possible allosteric effects, using AlloSigMA. We used our script to perform the submission and retrieval of the single point mutation scanning to and from the AlloSigMA server.
The allosteric effects on the sites (represented by the allosteric free energy ΔΔgsite) (substrate binding site or active site) was estimated from the residual responses as below:
ΔΔgres= ΔgUp-Mutation- ΔgDown-Mutation (1)
Where ΔΔgres is the scaled allosteric free energy change per residue representing the responses of each residue due to each single mutation. ΔgUp-Mutation and ΔgDown-Mutation are allosteric free energy changes with respect to events of UP-MUT (mutating to a bulky residue) and DOWN-MUT (mutating to a small residue) that are calculated and retrieved directly from the AlloSigMA webserver respectively.
The allosteric free energy change of the site, ΔΔgsite, is estimated by averaging over residues involved in the corresponding site (i.e. active site or substrate-binding site).
The submission script was executed for each residue in T4 Lysozyme protein to the AlloSigMA server (see Supplementary Video 1A for the demonstration of the automation process). The mutational event results from AlloSigMA were retrieved and further analyzed (Figure 2A).
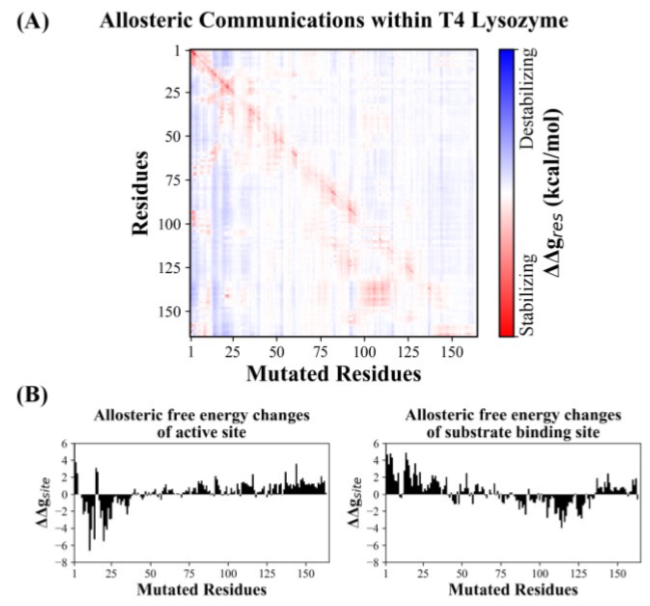
Figure 2: Analysis of allosteric communications in the T4 Lysozyme structure in the event of single point mutation using AlloSigMA.(A) The heatmap shows allosteric effect (destabilizing in blue and stabilizing in red) caused by single mutation (horizontal axis) on the protein structure. (B) Allosteric free energy changes,ΔΔgsite, of the active site (left) and the substrate binding site (right) due to single residue mutation event (horizontal axis).
We found different allosteric effects caused by the same set of residues on the active site and the substrate-binding site. Mutations occurring at regions of residues 1 to 25 appears to stabilize the active site (involving residues Glu11 and Asp20) while destabilizing the substrate binding site (involving residues Ser117 and Asn132), as shown in Figure 2B. However, we observed the opposite effect on the active site (e.g. destabilized) and the substrate-binding site (e.g. stabilized) when mutations occurred at regions of residues 80 to 130, including the mutation at P86 that was found previously (Alber et al., 1988). Nonetheless, the involvement of these two regions in any functional or regulatory roles have not yet been reported.
We documented the running time for the submission-retrieval process as described above (Table 1). For further comparison, we performed the script to perform the process of “UP then DOWN” mutation on other protein structures of various sizes and observed various average processing times required to perform the data submission and collection.
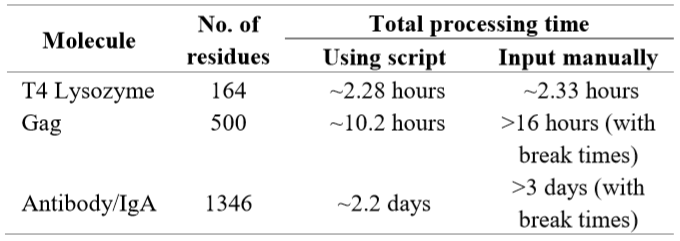
Table 1: Running time comparison between using the script and manual input for “UP then DOWN” Mutation.
We set out to alleviate some laborious and manual tasks of our lab projects using scripts, one of which involved using the AlloSigMA server for allosteric communication analysis. We presented a Python-based script that automates the process of submitting single mutation requests and retrieving results from the AlloSigMA server.
Our script maintains the accuracy and efficiency of the AlloSigMA server, while reducing the total processing time of the analysis for some large sized protein structures and with less room for human error that can arise from typos.
However, we observed that the processing time for each case was also affected by the load balance of AlloSigMA server when multiple users were performing the job on the server.
Our script initiates and promotes the convenience of scripting, especially using Python, in performing several submission and retrieval tasks to and from webservers. This highlights the scripting convenience complementary to the bioinformatics analyses.
The script could be provided upon request to the corresponding author.
Corresponding Author
# Email: phuasx@gmail.com.
SXP, KFC and CTTS drafted the manuscript. SXP and KFC designed the automated script. SXP and KFC prepared the figures, analysed the results and the video. CTTS supervised all aspects of the manuscript. SXP conceived the idea. All authors read and approved the manuscript.
Supplementary Video 1: Demonstration of the script running performing mutations on the AlloSigMA server. The demonstration was performed using the Firefox browser and the associated web browser engine. The printed outputs were displayed onto the terminal standard output. The video is available in this link:https://www.dropbox.com/s/dwgfaqosney1pmh/Figure%20S1.mp4?dl=0t. (17.9 MB)
We thank useful comments and feedback from the development team of the AlloSigMA webserver (Dr. Igor Berezovsky’s group in Bioinformatics Institute, A*STAR)
The authors declare no conflict of interest
Alber T, Bell J, Sun D, Nicholson H, Wozniak J, Cook S, Matthews B. Replacements of Pro86 in phage T4 lysozyme extend an alpha-helix but do not alter protein stability. Science 1988; 239: 631-635.
Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. Journal of molecular biology 1990; 215: 403-410.
Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, De Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E. ExPASy: SIB bioinformatics resource portal. Nucleic acids research 2012; 40: W597-W603.
Bairoch A, Boeckmann B. The SWISS-PROT protein sequence data bank. Nucleic acids research 1991; 19: 2247.
Bakker M, Post V, Langevin C D, Hughes J D, White J T, Starn J J, Fienen M N. Scripting MODFLOW model development using Python and FloPy. Groundwater 2016; 54: 733-739.
Chiang R Z-H, Gan S K-E, Su C T-T. A computational study for rational HIV-1 non-nucleoside reverse transcriptase inhibitor selection and the discovery of novel allosteric pockets for inhibitor design. Bioscience Reports 2018; 38: BSR20171113.
Guarnera E, Berezovsky I N. Structure-Based Statistical Mechanical Model Accounts for the Causality and Energetics of Allosteric Communication. PLOS Computational Biology 2016; 12: e1004678.
Guarnera E, Tan Z-W, Zheng Z, Berezovsky I N. AlloSigMA: allosteric signaling and mutation analysis server. Bioinformatics 2017; 33: 3996-3998.
Guido Python tutorial, Technical Report CS-R9526. 1995.
Henry V J, Bandrowski A E, Pepin A S, Gonzalez B J, Desfeux A. OMICtools: an informative directory for multi-omic data analysis. Database 2014; 2014.
Hunter J D. Matplotlib: A 2D graphics environment. Computing in science & engineering 2007; 9: 90-95.
Jones E, Oliphant T, Peterson P. SciPy: open source scientific tools for Python. 2014.
Kurochkin I V, Guarnera E, Berezovsky I N. Insulin-degrading enzyme in the fight against alzheimer’s disease. Trends in pharmacological sciences 2017.
Lehrer N. 2014. Which Is Easier To Learn, Java Or Python? Available from: https://www.hostgator.com/blog/easier-learn-java-python/.
McKinney W. Data structures for statistical computing in python. Proceedings of the 9th Python in Science Conference, 2010. Austin, TX, 51-56. 445
Oliphant T E. A guide to NumPy, Trelgol Publishing USA. 2006.
Oliphant T E. Python for scientific computing. Computing in Science & Engineering 2007; 9.
Shoichet B K, Baase W A, Kuroki R, Matthews B W. A relationship between protein stability and protein function. Proceedings of the National Academy of Sciences 1995; 92: 452-456.
Sinha N, Nussinov R. Point mutations and sequence variability in proteins: redistributions of preexisting populations. Proceedings of the National Academy of Sciences 2001; 98: 3139-3144.
Su C T-T, Kwoh C-K, Verma C S, Gan S K-E. Modeling the full length HIV-1 Gag polyprotein reveals the role of its p6 subunit in viral maturation and the effect of non-cleavage site mutations in protease drug resistance. Journal of Biomolecular Structure and Dynamics 2017: 1-12.
Su C T-T, Lua W-H, Ling W-L, Gan S K-E. Allosteric Effects between the Antibody Constant and Variable Regions: A Study of IgA Fc Mutations on Antigen Binding. Antibodies 2018; 7: 20.
Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids research 1994; 22: 4673-4680.
Zhao J, Nussinov R, Ma B. Allosteric control of antibody-prion recognition through oxidation of a disulfide bond between the CH and CL chains. Protein Eng., Des. and Sel. 2017; 30: 67-76.
Zhao J, Nussinov R, Ma B. Antigen Induced Dynamic Conformation Changes of Antibody to Facilitate Recognition of Fc Receptors. Biophysical Journal 2018; 114: 233a.

